Galaxy Training
Collection of training resources for Galaxy courses - Community Site
Proudly part of


PART I - Pre-processing and alignment
Step 1: Create a new (empty) history
Click on the history menu icon on the right column.
Select Create New and rename it sequence_align by clicking on the history name.
If the current history is already empty, just rename it.

Step 2: Import read files
Galaxy offers multiple solutions for getting data: local files, FTP, external repositories and data sharing. In this training session, we are going to import data using Shared Data
Select Data libraries from the Share Data menu as shown in the figure below.
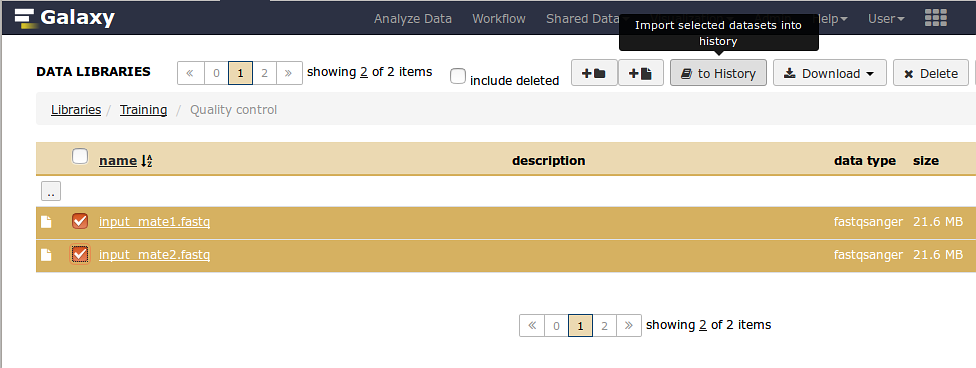
The files used during this session are contained into the Quality control folder inside the Training library
Expand the Quality control folder and select the files containing the paired reads:
- '’input_mate1.fastq’’
- '’input_mate2.fastq’’
Locate the import button to history at the top of the page and click it.
Click on Analyze Data to return to your workspace.
Step 3: Sequence quality check
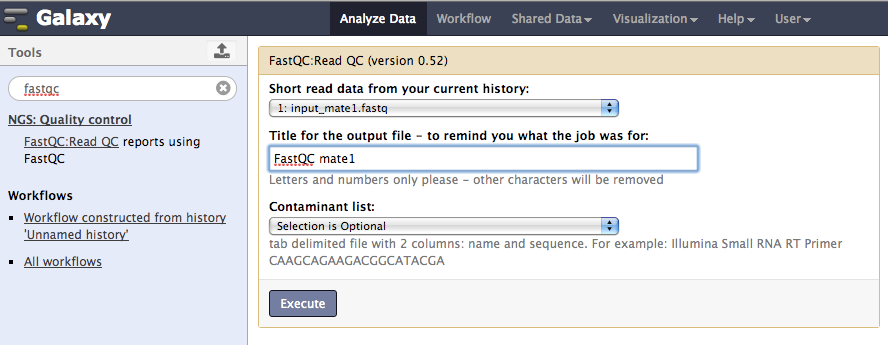
For each FastQ sequence, perform a quality check using FastQC
select the FastQC tool under NGS: Quality control menu, choose the desired FastQ file and execute the job
Step 4: Filtering and trimming
Analyze the FastQC results…

View the Per base sequence quality
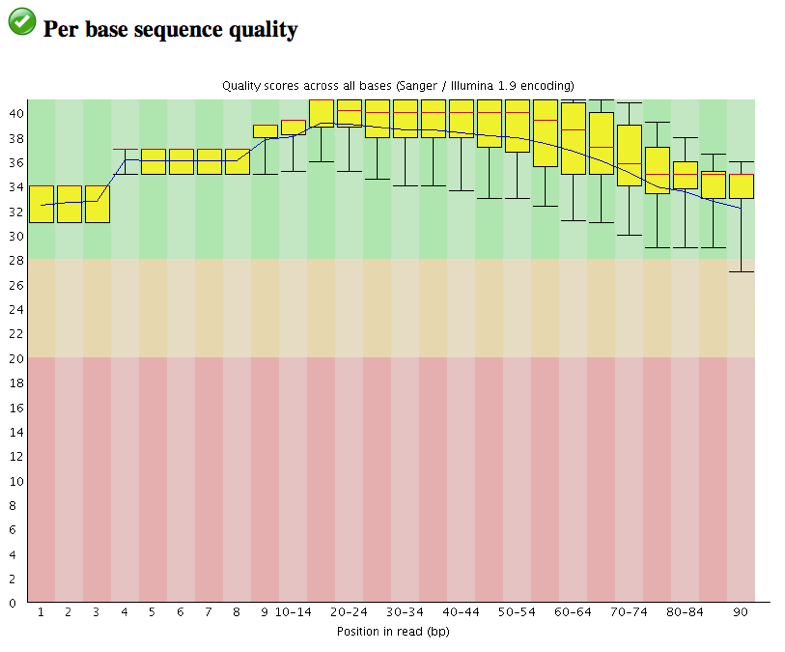
Trim the first 3 bases at 5’ and 3’ ends
Use the FASTQ positional and quality trimming tool in the NGS: Manipualtion menu to cut left/right sequence bases if they do not satisfy a minimal quality value (set by the user).
Select the paired-reads files and set the parameter values as in the following image
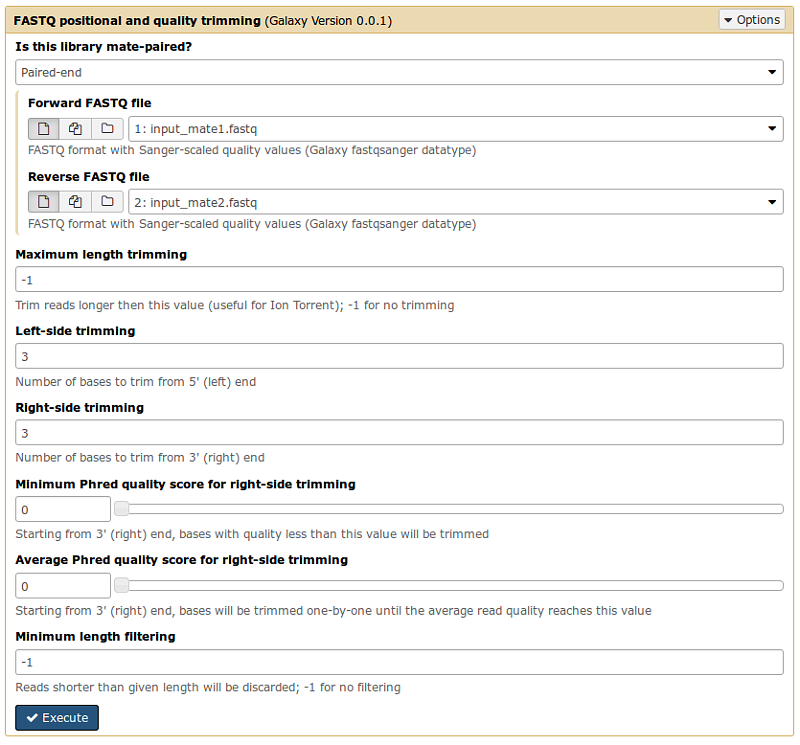
Tip: After trimming, if the sequence is shorter then a given length it’s removed from the the resulting FastQ file. Its mate sequence is removed too. Unpaired good sequences are kept in a separate file.
Step 5: Sequence alignment
We will use the BWA-MEM aligner to align the paired reads to the reference genome.
Mapping reads with BWA-MEM
The next step is the alignment of the processed reads to the reference genome using BWA, a fast software package for mapping low-divergent sequences against a large reference genome, such as human.
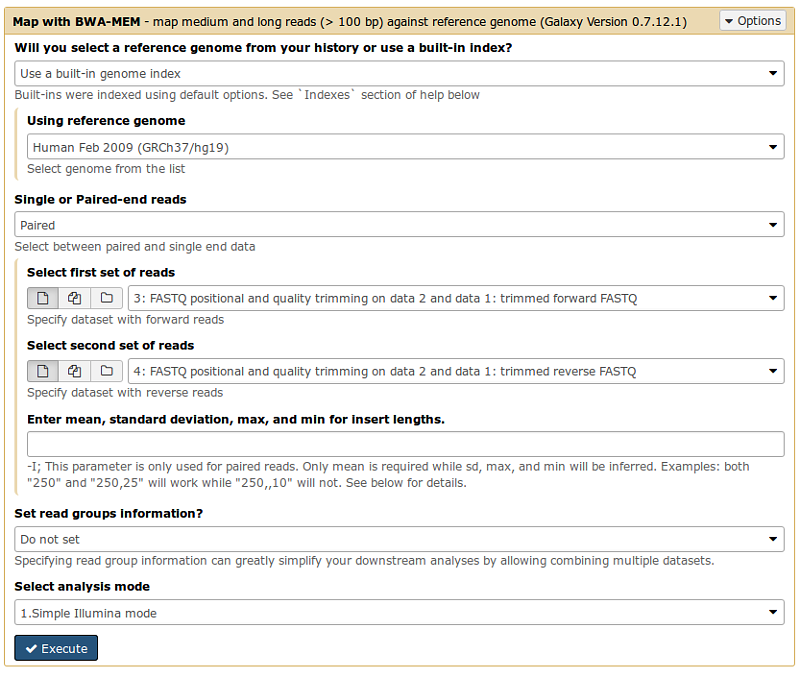
Select MAP with BWA-MEM tool from the NGS: Mapping menu
- Align the FASTQ files against the hg19 reference genome.
- Is this library mate-paired?: select ‘‘Paired ends’’ and choose the two filtered paired FastQ files
Step 6: Display BAM data using the UCSC Genome Browser
Select the bam output of Map with BWA-MEMM tool and choose the option Display at UCSC main.
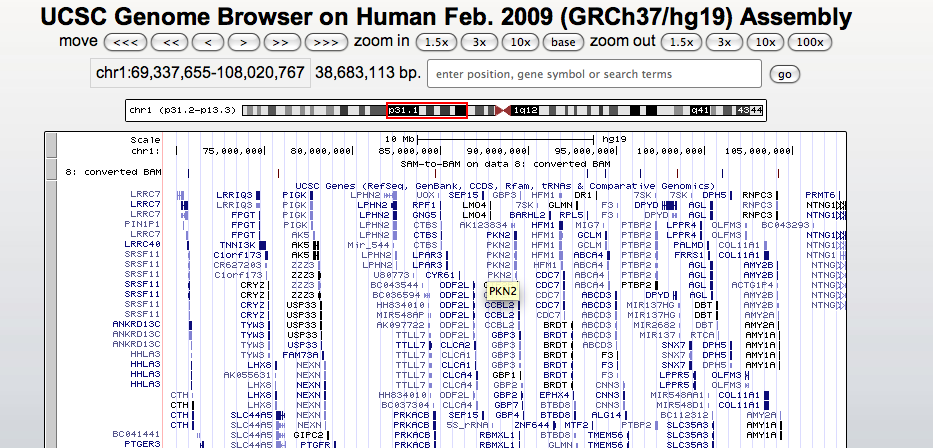
Example of UCSC Genome view
Jump on the genome to the PLK1S1 gene by typing its name in the box above the picture and check the coverage of the exons.
Tip: you can change the way your custom track is displayed by using the drop-down control under the section Custom tracks, just below the picture. Select the pack option.
Zoom-in at the level of a single exon and you should see the read pairs properly mapped linked by a black line.
Q: can you find reads aligned with mismatches (vertical red lines). Do you think they are sequencing errors or ‘‘real’’ variations in your sample?
Part II - Building and running a workflow
Step 7: Extract a workflow from history
Open the history menu and click on Extract Workflow
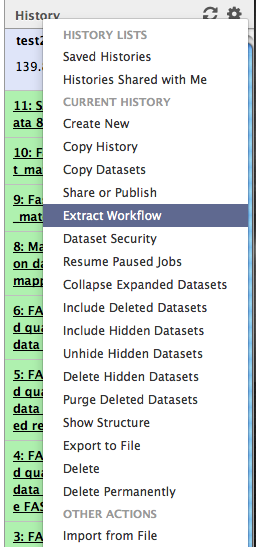
Rename your workflow “bwa_align” and click on  .
.

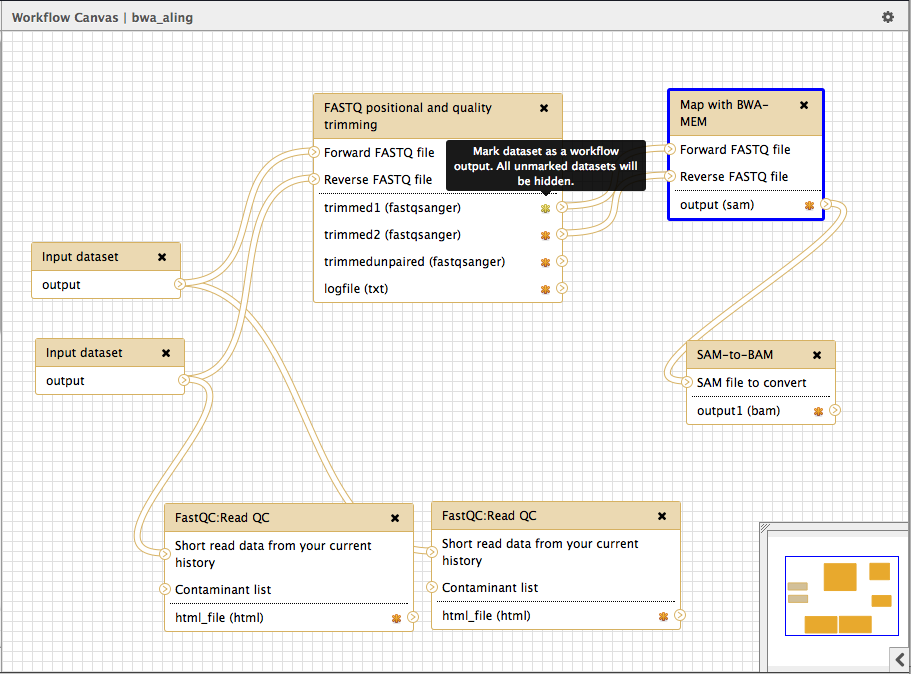
Step 8: Edit workflow
Select the Workflow tab in the galaxy menu bar.
Edit the “bwa_align” workflow.
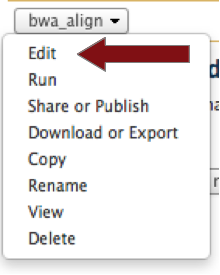
'’A graphical workflow editor will open’’
Here, you can modify all the program parameters and select the output files that will be displayed in your history by checking the proper check boxes.
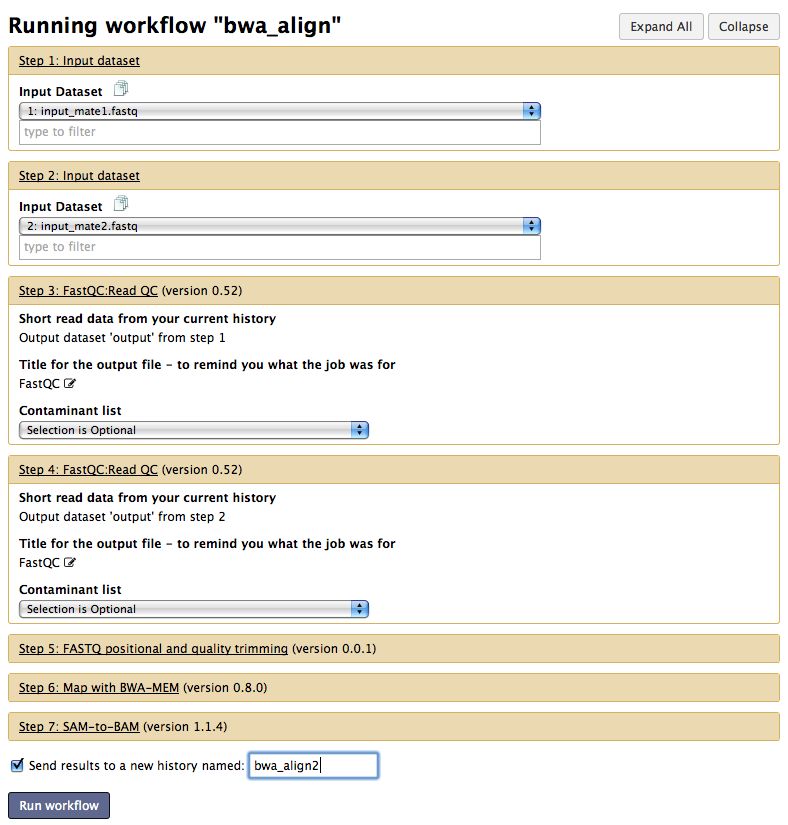
Save and Run your workflow
Now can save your wokflow and run it again (using the top right menu)
Run window